
Safety Assessment: Key Points Introduction of Extractables & Leachables
前幾期“濾”見新知系列我們介紹了關于除菌濾器的驗證概念及驗證主計劃和細菌截留測試,本篇主要介紹核心驗證項目之一:可提取物&浸出物以及安全性評估。
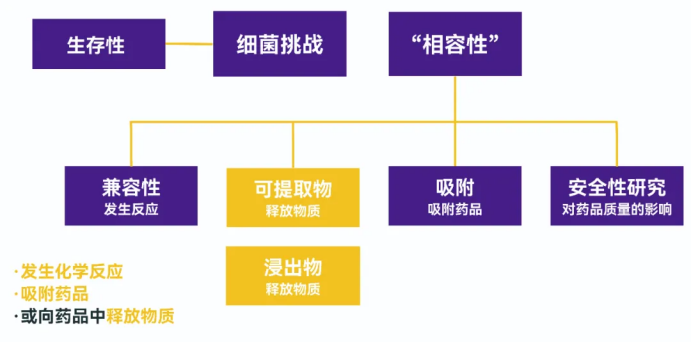
本文分為5個部分:
- 何謂可提取物?浸出物區別在哪?
- 雜質來源有哪些?受何因素影響?
- 如何開展可提取物/浸出物驗證?
- 驗證結果如何分析?
- 安全性評估怎么做?
何謂可提取物?浸出物區別在哪?
在NMPA的《除菌過濾技術及應用指南/2018》中有以下定義:
可提取物 -(Extractable)
在極端條件下(例如有機溶劑、極端高溫、離子強度、pH、接觸時間等),可以從過濾器及其它組件材料的工藝介質接觸表面提取出的化學物質。可提取物能夠表征大部分(但并非全部)在工藝介質中可能的潛在浸出物。
浸出物 -(Leachable)
在存儲或常規工藝條件下,從接觸產品或非接觸產品的材料中遷移進入藥物產品或工藝流體中的化學物質。浸出物可能是可提取物的一個子集,也可能包括可提取物的反應或降解后產物。
雜質來源有哪些?受何因素影響?
直接來源:濾器材質本身
濾膜:基礎材質(如PVDF、PES)及添加劑(增塑劑、抗氧劑、表面活性劑等)
其他組件:端蓋、濾殼、密封圈等非濾膜部件
間接來源:工藝條件影響
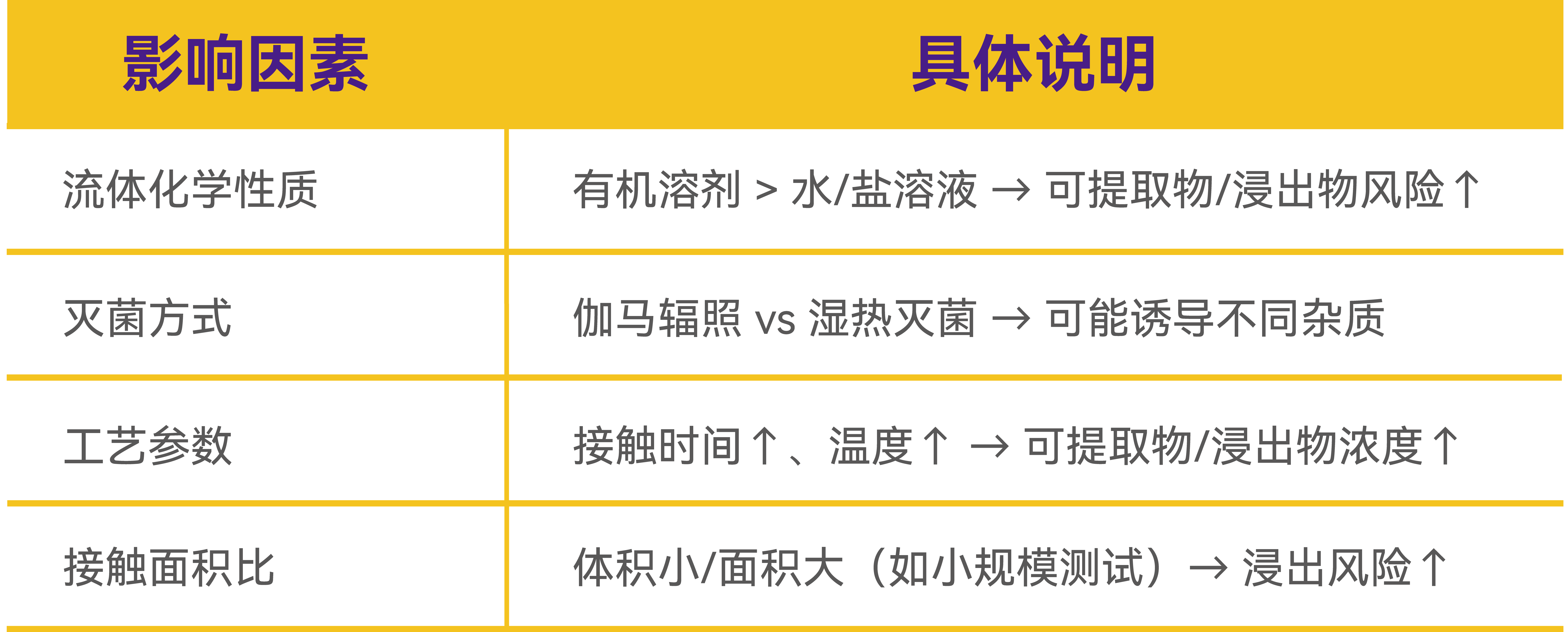

關鍵提醒:驗證需覆蓋整個過濾系統以及結合工藝條件考量,不局限于單一濾膜!
如何開展可提取物/浸出物驗證?
驗證之前先進行風險評估
“浸出物存在于最終原料藥和藥品中,通常包含在可提取物內,但由于分離和檢測方法的限制以及浸出物的量極小,很難被定量或定性。應先獲得最差條件下的可提取物數據,將其用于藥品的安全性評估。可提取物反映了浸出物的最大可能,無論是否要做浸出物試驗,可提取物的測試和評估都非常重要。”-- NMPA《除菌過濾技術及應用指南/2018》
從監管評估視角,可提取物數據不可缺少。 雖然浸出物直接反映實際應用場景(含背景干擾),且隨著檢測能力提升更具可操作性,CED指南特別強調:可提取物代表浸出物的理論最大暴露風險(最差條件)。二者實為互補而非替代——可提取物劃出安全閾值邊界,浸出物確認日常水平,共同構成風險評估體系。
可提取物的風險評估考量
可提取物實驗前需做風險評估,非所有過濾工藝都需復雜驗證,關鍵評估維度如下表:


可提取物的風險評估-關鍵評估維度
基于風險評估的可提取物驗證試驗
根據USP 665建議,不同風險等級的驗證實驗主要分為兩類:
1. 生物反應性測試
低風險豁免|中風險測細胞毒性|高風險增加體內測試
2. 可提取物實驗
·模型溶劑選擇:低風險與中風險樣品均使用50%乙醇水溶液;高風險樣品需額外增加pH=3的酸溶液和pH=10的堿溶液。
·分析內容分層級:
A.低風險&中風險:基礎測試包括NVR(非揮發性殘留物)和UV吸收。
B.中風險:在基礎測試上增加有機提取物測試。
C.高風險:需結合多種溶劑(50%乙醇、酸、堿溶液)提取物,進行綜合測試。
·分析方法擴展:除低/中風險的分析項目外,高風險測試中必要時需補充元素測試。
需要注意的是,上述方案主要適用于常規工藝場景,無法覆蓋所有情況。例如:
-有機相濃度>50%:此時采用50%乙醇水溶液進行提取可能不再適用;
-極端pH(>10 或 <3)溶液。
針對此類情況,我們需根據實際工藝條件,定制化設計實驗方案,確保可提取物研究更精準、更全面地滿足法規要求。
驗證結果如何分析?
可提取物和浸出物的檢測方法包括定量和定性兩類,如 LC/PDA/MS, DI-GC/MS, HS-GC/MS, ICP/MS, UV, FTIR, NVR, TOC,為了保證分析方法的可靠性,需對分析方法進行驗證或確認。選擇哪幾種分析方法,取決于實際的藥品和生產工藝以及過濾器生產商對過濾器的充分研究。
具體分析方法詳見下表:

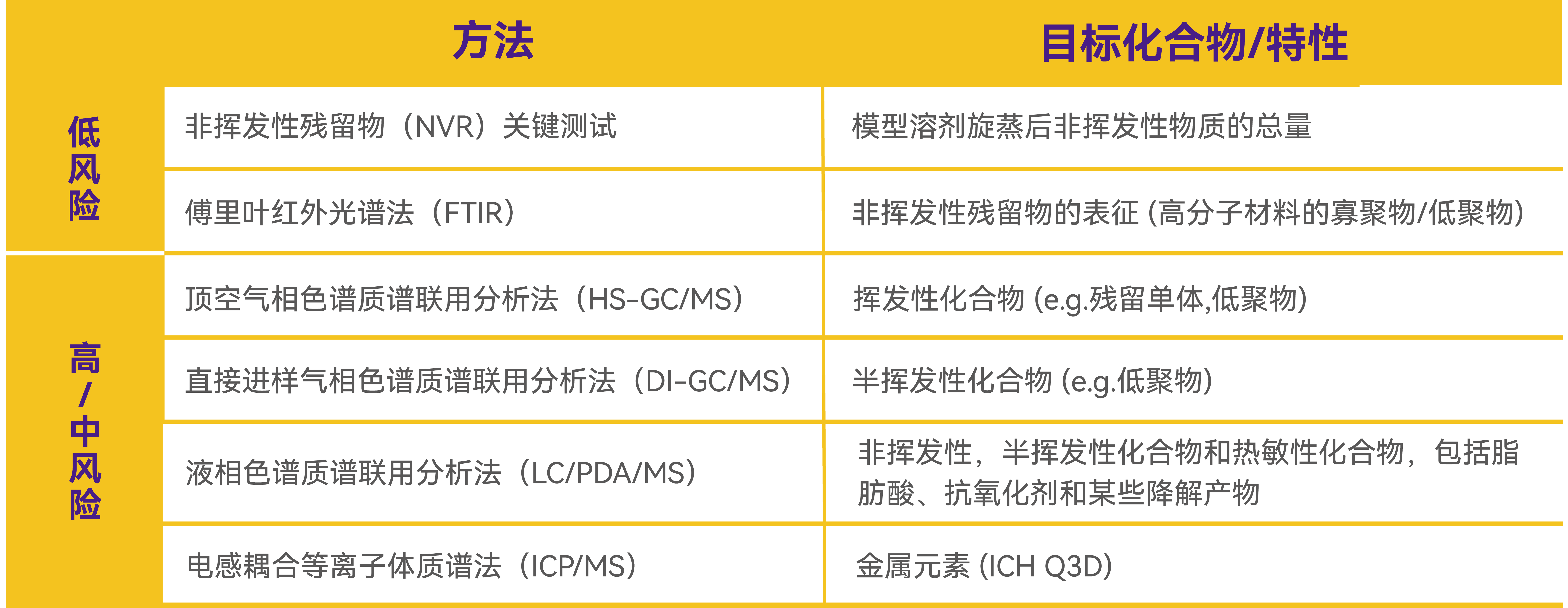
基礎 & 完整可提取物分析方法
浸出物試驗研究
浸出物實驗通常在可提取物的安全性評估顯示其對患者風險不可接受時啟動。
進行浸出物研究前,需執行系統適用性研究,此步驟比較重要,用以確認分析方法的可行性,克服在實際藥品中可能存在的干擾物影響。
其次,是浸出物的分析:
1.基于可提取物數據預測潛在目標浸出物
2.優先關注:
·可提取物研究中檢出量高的物質
·檢測靈敏度較低的物質(避免漏檢)
·高毒性物質(即使含量低也可能風險顯著)
安全性評估怎么做?
為什么要開展安全性評估
- 基于安全方面的考慮和監管部門的要求
“在完成可提取物或浸出物試驗后,應針對過濾器可提取物或浸出物的種類和含量,結合藥品最終劑型中的濃度、劑量大小、給藥時間、給藥途徑等對結果進行安全性評估,以評估可提取物和浸出物是否存在安全性風險。” --《除菌過濾技術及應用指南》,NMPA, 2018年10月
安全性評估流程
完成可提取物和浸出物研究后,需對檢出的雜質進行系統性安全性評估。其核心步驟如下:
1. 雜質分析與暴露量計算
雜質分析:通過可提取物/浸出物研究明確雜質的種類和定量水平。
暴露量計算:結合藥品的臨床使用參數(如劑量、劑型、給藥周期),計算患者對雜質的每日暴露總量(Total Daily Exposure)。
2. 毒理學評估
評估執行:由具備資質的毒理學家根據暴露量數據開展安全性評估。
評估方法:
A.毒理學關注閾值(TTC, Threshold of Toxicological Concern):適用于缺乏特定毒理學數據的化合物。
B.允許每日暴露量(PDE, Permitted Daily Exposure):適用于具備充分毒理學數據的化合物。
方法選擇:TTC或PDE的適用性需根據物質特性與數據完整性科學判定。
3. 風險判定與風險控制
可接受風險:若暴露量 < TTC或PDE值 → 風險可忽略,過濾器可安全用于生產。
不可接受風險:若暴露量 > TTC或PDE值 → 需啟動風險控制措施:
A.工藝優化:增加稀釋因子、調整工藝參數降低雜質水平;
B.分析升級:采用高靈敏度方法(如GC-MS/LC-MS)重新檢測;
C.再評估:控制措施實施后,重新執行安全性評估。

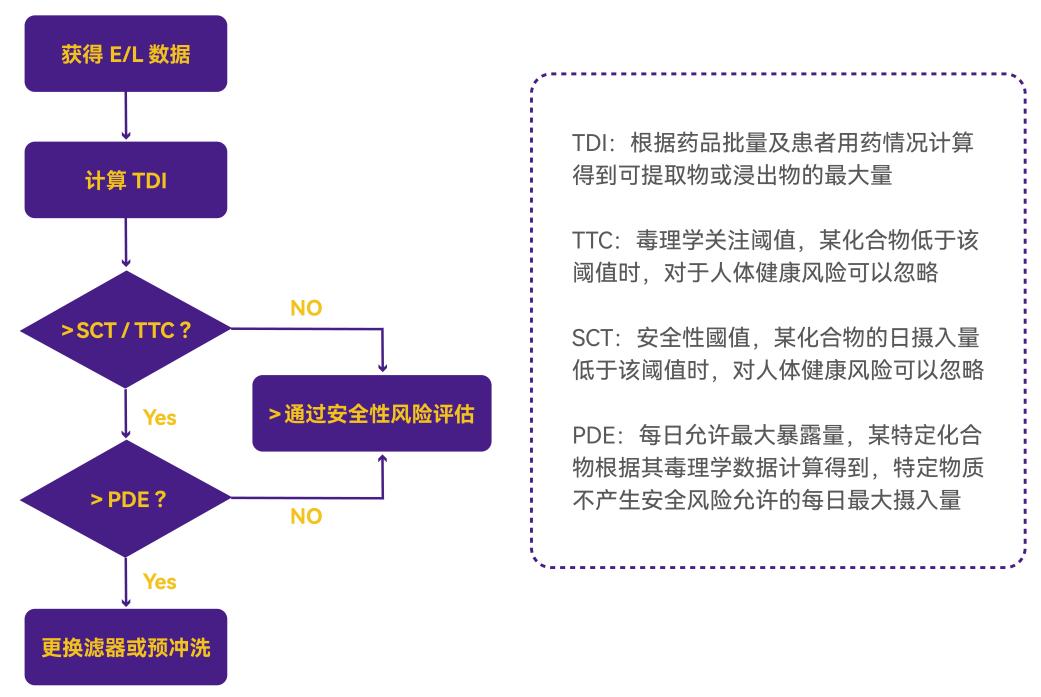
基于過程的安全性評估策略總結
1. 風險初評
根據過濾器配置、工藝條件等設計可提取物研究方案。
2. 安全決策樹
·風險可忽略(可提取物數據支持):直接提交監管申報。
·風險不可忽略:執行浸出物定量分析;或實施風險控制措施后重新評估。
3. 監管提交
最終確認風險可控,提交完整證據鏈至監管機構。
總結
本期要點聚焦過濾系統安全性評估的核心——可提取物與浸出物,可提取物(最差條件下提取的化學物質)和浸出物(實際工藝下遷移的雜質)是評估過濾系統安全性的關鍵,需基于風險等級(溶液性質、工藝條件、制品風險)選擇模型溶劑(基礎:50%乙醇;高風險+酸堿液)和分析內容(基礎:NVR+UV;升高:有機物+元素)。最終通過計算患者每日暴露量,比照TTC/PDE毒理學標準進行安全性判定,確保藥用安全。
參考法規與指南
[1]《藥品生產質量管理規范》(2010年修訂)。
[2]《除菌過濾技術及應用指南》(國家藥品監督管理局2018年第85號)。
[3] Bio-Process System Alliance (BPSA) Recommendations for Testing and Evaluation of Extractables from Single-Use Process Equipment, 2010.
[4] USP PF <665>, Plastic Materials, Components, and Systems Used in the Manufacturing of Pharmaceutical Drug Products and Biopharmaceutical Drug Substances and Product.
[5] BPOG: Best Practices Guide For Extractables Testing of Polymeric Single-Use Components Used in Biopharmaceutical Manufacturing, 2020.
[6] ICH HARMONISED GUIDELINE: Assessment and Control of DNA Reactive (Mutagenic) Impurities In Pharmaceuticals To Limit Potential Carcinogenic Risk M7 (R1).
[7]《化學藥品注射劑生產所用的塑料組件系統相容性研究技術指南》(征求意見稿),2020。
About Us
Products
Application
Service
News
Careers
Contact Us
400-626-3749
